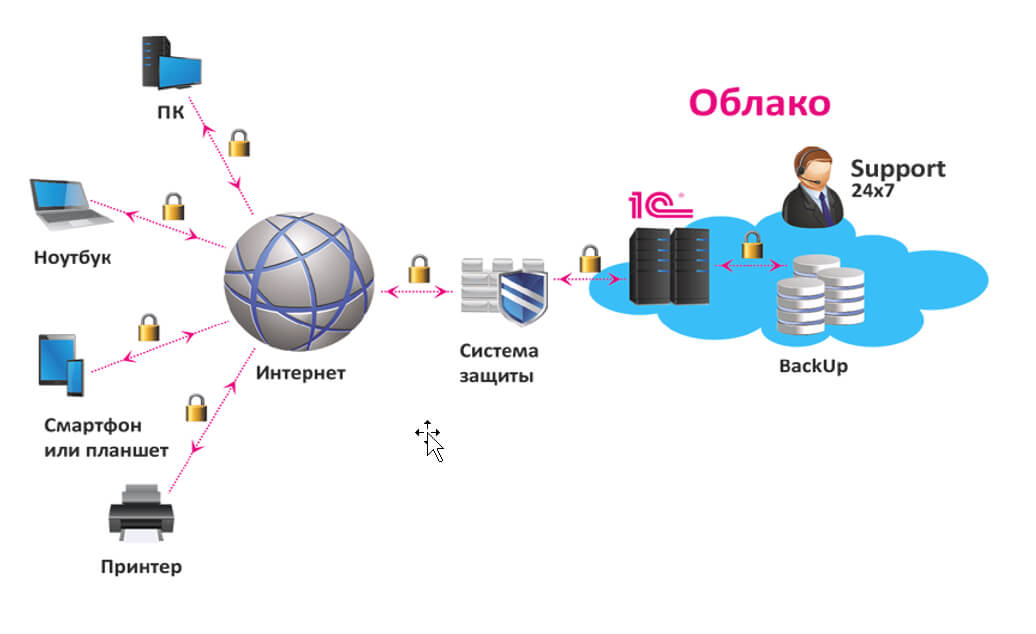
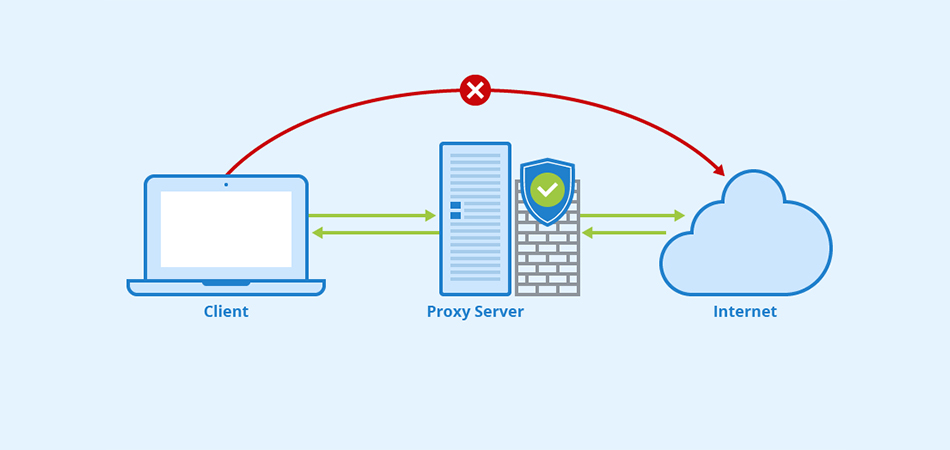
Выделенный сервер — это мощное и эффективное решение для бизнеса и крупных проектов в Интернете, требующих высокой производительности, безопасности и стабильности. Рассмотрим подробнее, что такое выделенный сервер Windows, его преимущества и возможные сценарии использования. Что такое выделенный сервер Windows? Выделенный сервер Windows — это физический сервер, который хостинг-провайдер предоставляет в аренду одному клиенту. Это обеспечивает […]